Team
In den Zellen aller Organismen steuern Proteine fast jeden zellulären Prozess, vom Zellwachstum und der Zellteilung bis hin zur Stoffwechselaktivität, Zellmotilität oder Signalübertragung. Daher ist der Prozess der Proteinsynthese von zentraler Bedeutung für das reibungslose Funktionieren der Zellen. Wir entwickeln theoretische Beschreibungen und Algorithmen zur Beschreibung und Analyse des Prozesses der Proteinbiosynthese in verschiedenen Arten von Organismen und künstlichen Expressionssystemen. Um unsere Modelle zu entwerfen und zu parametrisieren, arbeiten wir eng mit Biochemie- und Biologielabors zusammen. Wir fördern offene und reproduzierbare Wissenschaft.
Translation in Chloroplasten
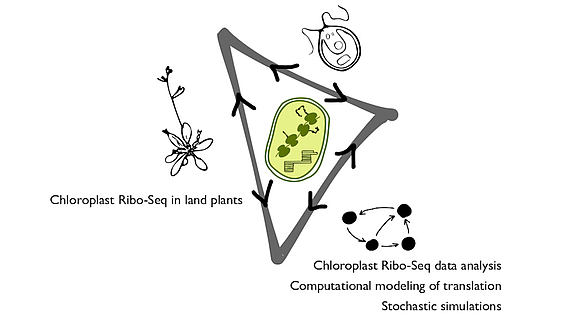
Chloroplasten sind pflanzliche Zellorganellen, die für das Leben auf der Erde unerlässlich sind. Sie wandeln über den Prozess der Photosynthese Lichtenergie in biochemische Energie um. Im Rahmen des DFG-Projekts Vergleichende, quantitative Untersuchung der Dynamiken des chloroplastidären Translationsapparates in einer Grünalge und in Landpflanzen (Projektnummer 437345987) in Zusammenarbeit mit Prof. Dr. Felix Willmund an der Universität Kaiserslautern und Dr. Reimo Zoschke am Max-Planck-Institut für molekulare Pflanzenphysiologie untersuchen wir die Translation in Chloroplasten.
Wir nutzen unter anderem Ribosome Profiling (Ribo-Seq) Datensätze, um das Translationsverhalten in verschiedenen Modellsystemen zu analysieren, die Häufigkeit von Frame-Shifting und ribosomalem Drop-off zu quantifizieren und kinetische Informationen des Translationsprozesses aufzudecken. Die gesammelten Ergebnisse werden verwendet, um ein Computermodell der Translation in Chloroplasten zu entwerfen und zu parametrisieren. Mit diesem Modell können wir die Proteinsynthese in Chloroplasten weiter untersuchen, zum Beispiel die Robustheit und Anpassungsfähigkeit der Translation bei Störungen durch Mutationen, Translationspausen oder den Einfluss von Codon-Anticodon-Superwobbling auf die Elongationsgeschwindigkeit und -genauigkeit.
Analyse von Sequenzdatensätzen
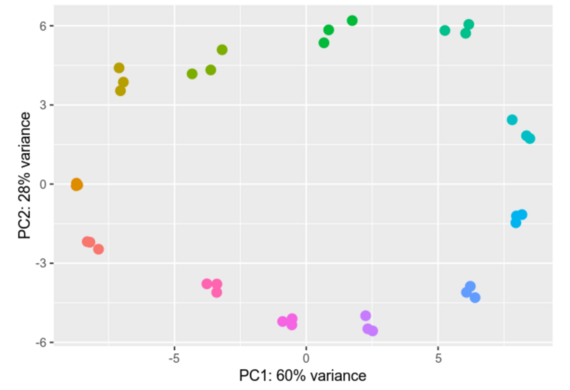
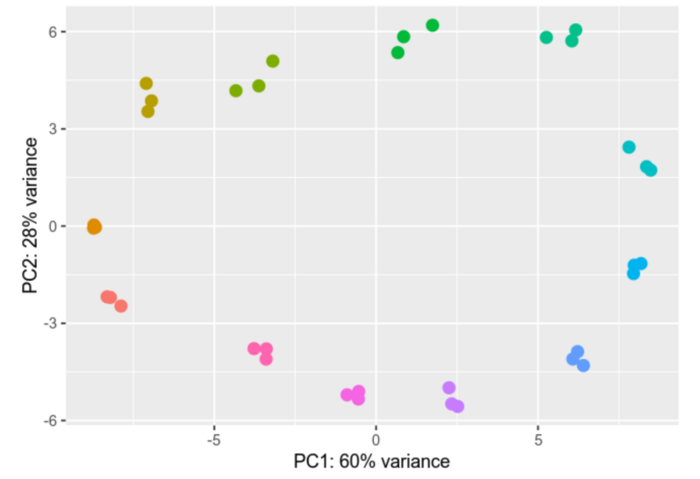
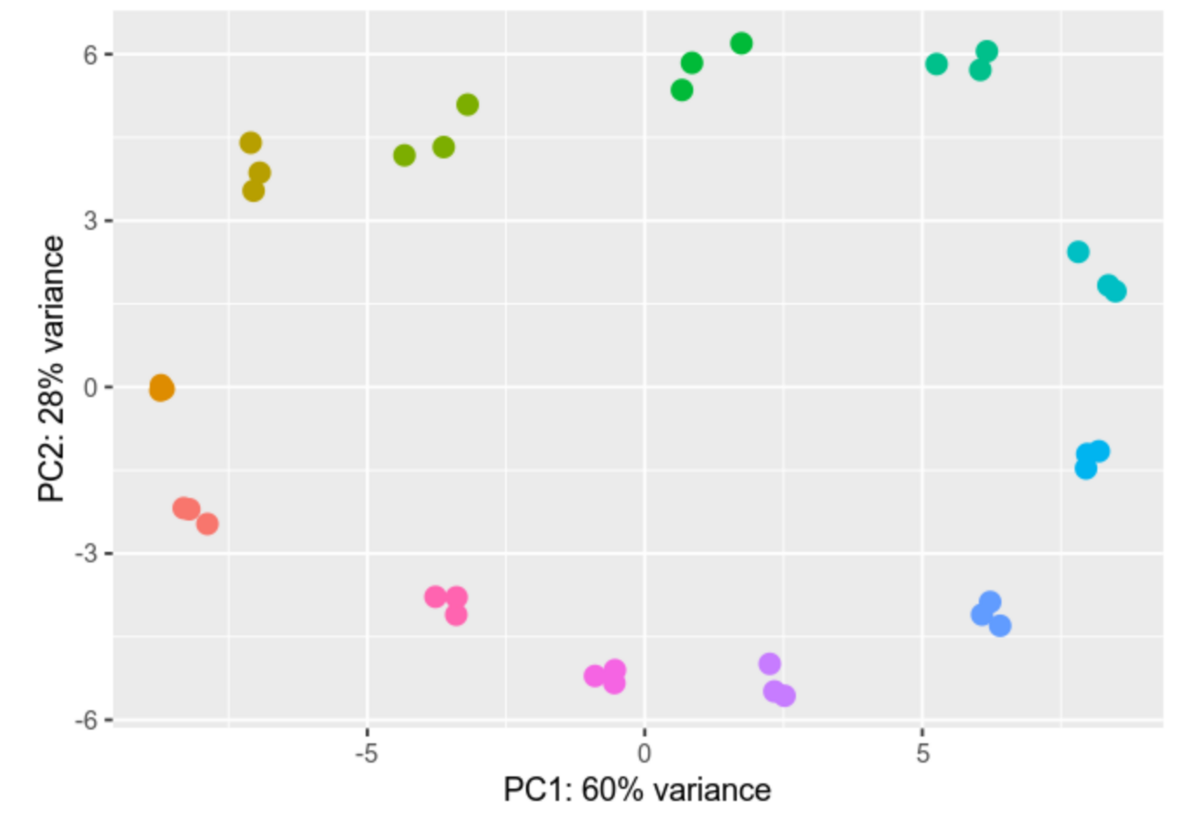
Die differentielle Genexpressionanalyse ist ein Prozess, der darauf abzielt, das komplexe Netzwerk von Genen und ihre unterschiedlichen Aktivitätsniveaus in verschiedenen Proben oder unter unterschiedlichen Bedingungen zu entschlüsseln. Wir vergleichen die Genaktivität zwischen verschiedenen Zelltypen, Zuständen oder Behandlungen und untersuchen die Feinheiten ihrer Aktivierung oder Deaktivierung, um den Umfang dieser Veränderungen zu messen.
Diese Art der Analyse ist besonders wichtig um zu verstehen, wie Zellen auf externe Einflüsse reagieren und welche biologischen Prozesse beeinflusst werden. In der Praxis kann die differentielle Genexpressionanalyse für eine Vielzahl von Anwendungen genutzt werden, wie beispielsweise in der Erforschung von Krankheiten, der Entwicklung neuer Medikamente und der Verbesserung von Therapien.
In unserer Arbeitsgruppe analysieren wir Ribosome Profiling (Ribo-Seq) und RNAseq-Datensätze unter der Verwendung der Software-Packete DESeq2 und EdgeR. Die Ergebnisse unserer Analysen werden durch visuelle Darstellungen veranschaulicht, darunter Heatmaps und Hauptkomponentenanalysen (PCAs), die die feinen Unterschiede in den Genexpressionsprofilen veranschaulichen.
Proteinkomplexbildung
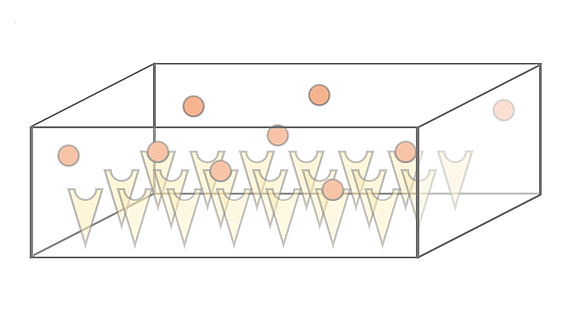

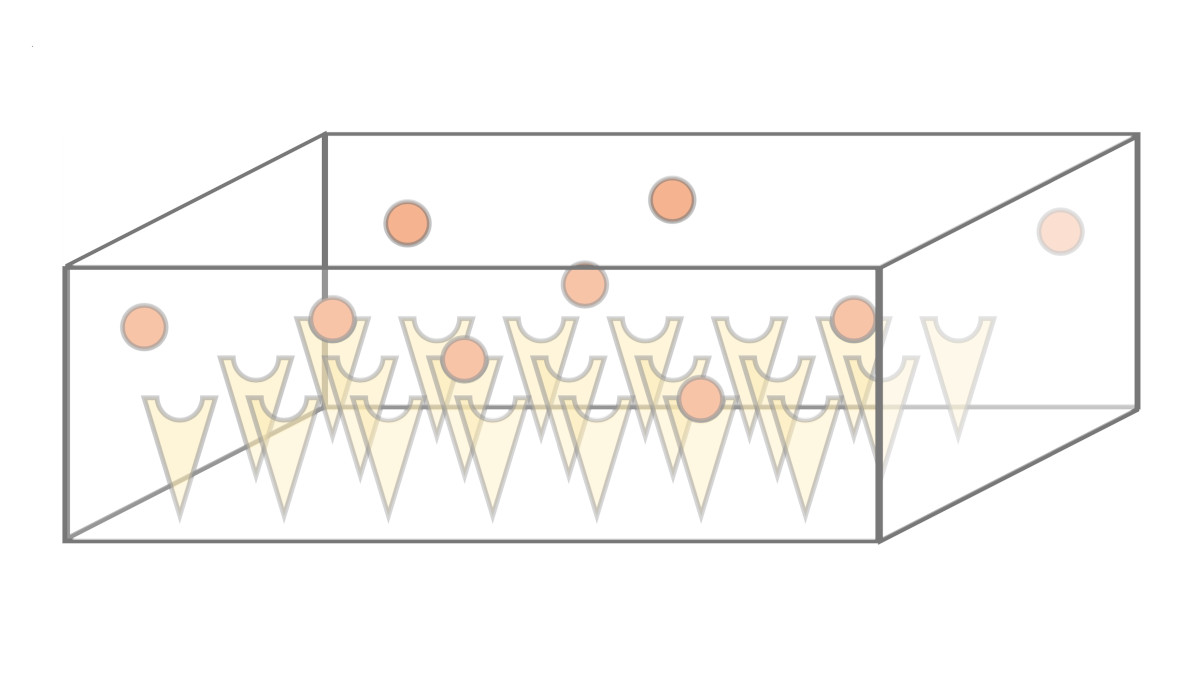
Wir untersuchen die Dynamik und Effizienz der Proteinkomplexbildung in quasi-zweidimensionalen, siliziumbasierten Kompartiment-Expressionssystemen, die vom Bar-Ziv-Labor (Weizmann Institut, Israel) eingeführt wurden. In diesen Systemen werden einzelne Proteinuntereinheiten in lokal begrenzten Domänen synthetisiert und durch molekulare Fallen auf der Kompartmentoberfläche eingefangen. Wir untersuchen systematisch, wie die Komplexbildung und die Protein-Fallen-Bindung von den Systemparametern, wie Syntheserate, Diffusionskonstante und Fallen-Bindungsaffinität der exprimierten Proteinuntereinheiten, beeinflusst werden.
Reaktions-Diffuisons-Systeme mit konkurrierenden Reaktionen
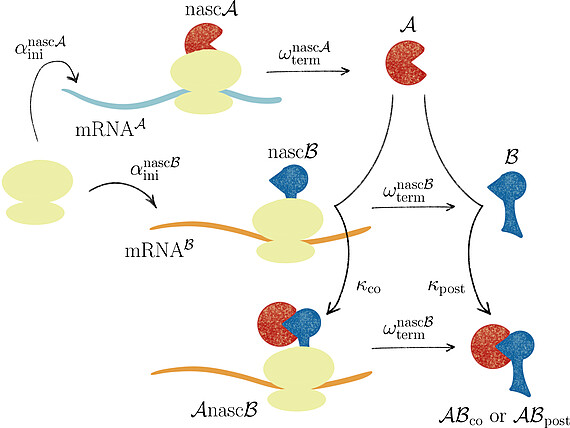
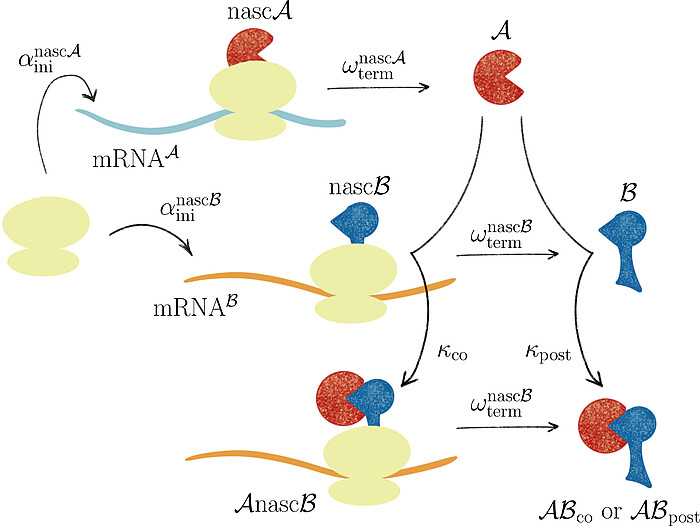
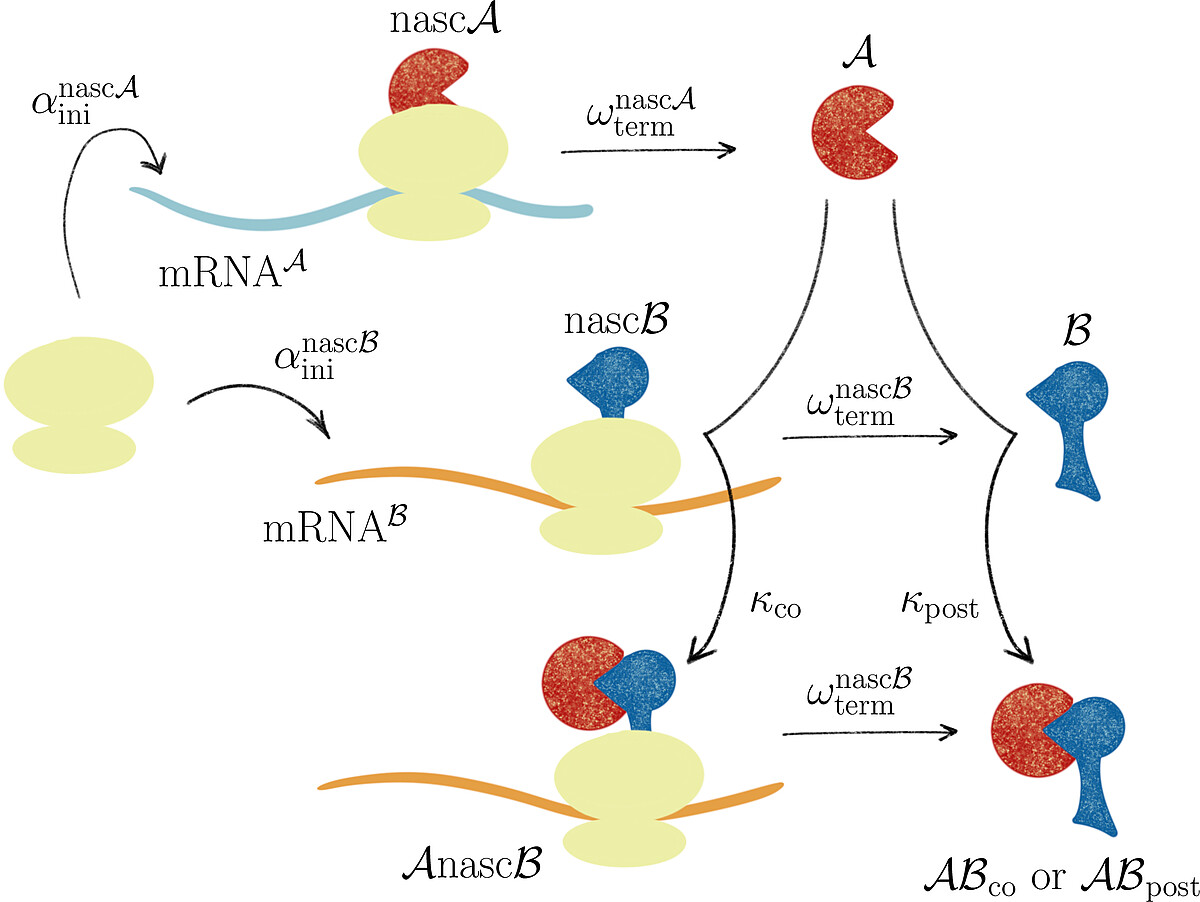
Gemeinsam mit Prof. Johannes Lankeit (Fakultät für Mathematik und Physik, LUH) haben wir für die zelluläre Proteinbiosynthese das Konzept der asymmetrischen, konkurrierenden Reaktionen eingeführt, um Dominanzen verschiedener Reaktionspfade bei der ko- und posttranslationalen Protein-Komplex-Assemblierung zu bestimmen. Unsere Analyse ergab, dass das Langzeitverhalten eines Translationssystems ausschließlich durch das Produktionsratenverhältnis der beiden Proteinuntereinheiten bestimmt wird, unabhängig von den Bindungskonstanten für die post- und ko-translationale Assemblierung.
Optimierung der Proteinexpression



Die Redundanz des universellen genetischen Codes, bei dem 61 verschiedene Codons für nur 20 proteinogene Aminosäuren codieren, ermöglicht synonyme Mutationen, die das Genprodukt nicht verändern. Jedes Codon zeichnet sich durch unterschiedliche physikalisch-chemische Eigenschaften aus, die z. B. seine Neigung zur Bildung von mRNA-Sekundärstrukturen und seine Decodierungsgenauigkeit beeinflussen. Folglich werden verschiedene Codons von den übersetzenden Ribosomen unterschiedlich schnell verarbeitet. Wir haben die Software OCTOPOS entwickelt, ein Werkzeug zur computergestützten Codon-Optimierung, das synonyme Sequenzen für eine verbesserte heterologe Proteinexpression auf der Grundlage einer Vielzahl von mRNA-spezifischen Eigenschaften vorschlägt. OCTOPOS integriert drei Ebenen der Modellierung:
- ein umfassendes, mechanistisches mathematisches Modell der in-vivo mRNA-Translation, das eine Vielzahl von molekularen Interaktionswegen und deren Kinetik berücksichtigt
- stochastische Computersimulationen, die den vorzeitigen Ribosomenabfall, die mRNA-Stabilität und die Bildung von Ribosomenstau erfassen
- einen Algorithmus für maschinelles Lernen, der anhand großer Genexpressionsdatensätze organismusspezifisch trainiert wird, um die Bedeutung weiterer mRNA-spezifischer Eigenschaften zu berücksichtigen und zu bewerten.
Stochastische Modellierung der Proteinsynthese
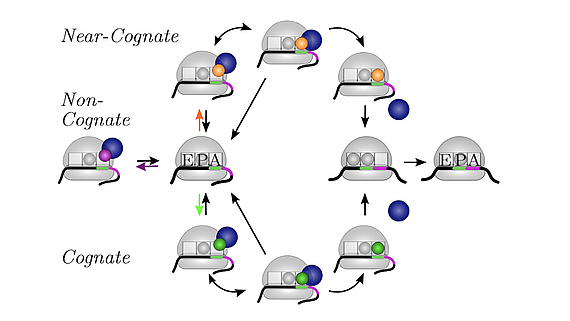
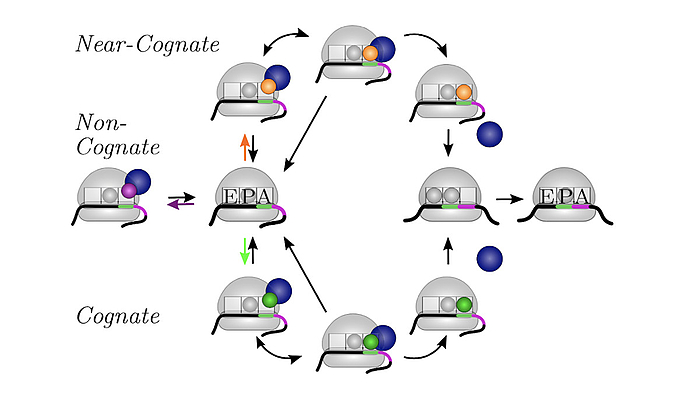
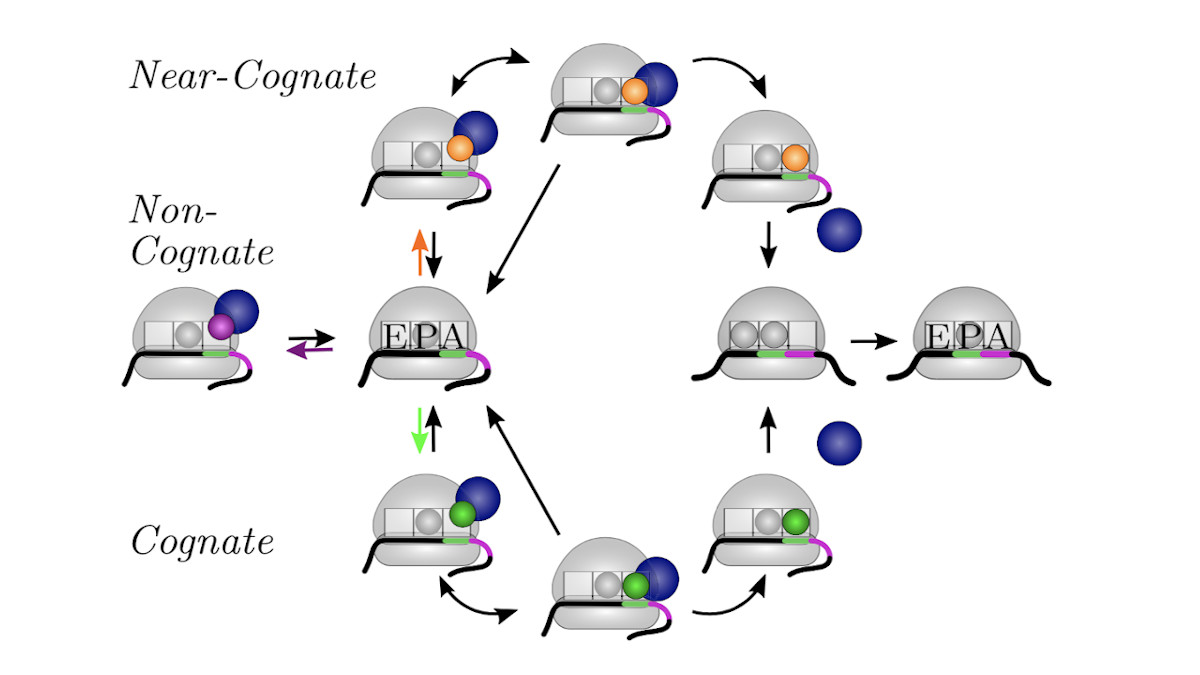
Wir entwickeln und verwenden stochastische Beschreibungen der zellulären Proteinsynthese um zu verstehen, wie die Geschwindigkeit und die Zuverlässigkeit des Prozesses bestimmt werden. Darüber hinaus haben wir eine Methode entwickelt, um die Kinetik der Proteinsynthese in der Zelle aus in-vitro Messungen abzuleiten und das in-vivo Modell zu verwenden, um die Auswirkungen von Störungen zu bestimmen. So haben wir beispielsweise untersucht, unter welchen Bedingungen eine milde Hemmung des Elongationsfaktors EF-Tu die Proteinsynthese stoppt.
Stochastische Modellierung der Translation in humanen Mitochondrien
Mitochondrien sind Organellen, in denen die Zellatmung stattfindet. Sie besitzen ihr eigenes Genom und einen Translationsapparat. Wir wollen die Translation in humanen Mitochondrien mit Hilfe unseres statistischen Modells beschreiben und den Einfluss verschiedener Mutationen im mitochondrialen Genom auf die Proteinsynthese untersuchen. Dadurch sollen die Mechanismen hinter verschiedenen Mitochondriopathien besser verstanden werden.
Modellierung von photosynthetischen State Transitions
State Transitions spielen eine wichtige Rolle bei der Regulation der Photosynthese unter schwachem Licht bei höheren Landpflanzen. Lichtsammelkomplexe können zwischen den Photosystemen migrieren, um die Absorptionsenergie auszugleichen. Basierend auf der Arbeit von Matuszyńska et al. beschreiben wir den Mechanismus von State Transitions mit einem System von Differentialgleichungen. Die Ergebnisse unserer Simulationen werden mit experimentellen Daten der Arbeitsgruppe Pflanzenphysiologie (Institut für Botanik, LUH) verglichen um das derzeitige Verständnis des Mechanismus von State Transitions zu verbessern.
Positionsabhängige Translationskinetik
Ribosomen bestehen aus einer kleinen und einer großen Untereinheit. Während der Proteinsynthese durchquert die naszierende Peptidkette den ribosomalen Tunnel, einen internen Tunnel in der großen Untereinheit. Um die Wechselwirkungen zwischen dem Tunnel und dem naszierenden Peptid sowie deren Auswirkungen auf den Syntheseprozess zu untersuchen, wurde in Zusammenarbeit mit dem Rodnina-Labor am Max-Planck-Institut für biophysikalische Chemie in Göttingen eine in-vitro-Methode zur Überwachung der cotranslationalen Bewegung entwickelt. Ein Fluorophor wurde an der naszierenden Peptidkette befestigt und das Fluoreszenzsignal zeitaufgelöst detektiert. Cotranslationale Ereignisse (z.B. Peptidfaltung) sowie die Struktur und chemische Umgebung des Ausgangstunnels beeinflussen das Fluoreszenzsignal der übersetzten mRNA, was zu einer eindeutigen, zeitabhängigen Fluoreszenzsignatur für diese Sequenz führt.
Das Experiment wird als Ensemble-Experiment durchgeführt und die einzelnen Fluoreszenzsignaturen überlagern sich zu einer Gesamtfluoreszenzsignatur, die zerlegt werden muss, um die zugrundeliegende Information aufzudecken. Um Erkenntnisse über die positionsabhängigen Intensitäten und die Kinetik des Prozesses zu gewinnen, modellieren wir die Translation, inklusive der detaillierten biologischen Teilprozesse, als Markov-Prozess. Mit dieser Methode entdeckten wir, dass sich die Translation von Poly(U)-mRNAs am vierten UUU-Codon dramatisch verlangsamt.
Unsere Methode bietet einen neuartigen Ansatz zur Untersuchung der Translationskinetik in Ensemble-Experimenten und kann für die Untersuchung anderer biochemischer Prozesse, die von prozessiven Enzymen angetrieben werden, erweitert werden. Ein Hauptanliegen unserer Arbeit ist es, eine sinnvolle rechnerische Analyse der bereitgestellten Daten zu finden und eine Überanpassung zu vermeiden. Insbesondere bei hochkomplexen Prozessen muss die Anzahl der Parameter im Modell sorgfältig definiert werden. Einerseits muss die Komplexität des biochemischen Prozesses widergespiegelt werden, andererseits müssen die Ergebnisse eindeutig und zuverlässig sein.
Kooperationen
Kontakt


Prof. Dr. Sophia Rudorf
Telefon
E-Mail
Adresse
115/4160, Herrenhäuser Str. 2
30419 Hannover
30419 Hannover
Prof. Dr. Sophia Rudorf
Telefon
E-Mail
Adresse
115/4160, Herrenhäuser Str. 2
30419 Hannover
30419 Hannover





