Meet our team
In cells of all organisms, proteins govern almost every cellular process from cell growth and division to metabolic activity, cell motility or signaling. Thus, the process of protein synthesis is pivotal for a proper functioning of the cells. We develop theoretical frameworks and algorithms to describe and analyze the process of protein biosynthesis in different kinds of organisms and artificial expression systems. To design and parameterize our models, we closely collaborate with biochemistry and biology labs. We encourage open and reproducible science.
In cells of all organisms, proteins govern almost every cellular process from cell growth and division to metabolic activity, cell motility or signaling. Thus, the process of protein synthesis is pivotal for a proper functioning of the cells. We develop theoretical frameworks and algorithms to describe and analyze the process of protein biosynthesis in different kinds of organisms and artificial expression systems. To design and parameterize our models, we closely collaborate with biochemistry and biology labs. We encourage open and reproducible science.
Translation in Chloroplasts
Chloroplasts are plant cell organelles that are essential for life on Earth. They convert light energy into biochemical energy via the process of photosynthesis. As part of the DFG project Comparative quantitative analysis of the chloroplast translation apparatus and its dynamic behavior in a green alga and land plants in cooperation with Prof. Dr. Felix Willmund at the University Kaiserslautern and Dr. Reimo Zoschke at the Max Planck Institute of Molecular Plant Physiology we investigate translation in chloroplasts.
We use a variety of data sets to analyse the translation behaviour in different chloroplast model systems, to quantify the frequencies of frame-shifting and ribosomal drop-off and to reveal kinetic information of the translation process. The gathered results are used to design and parameterize a computational model of translation in chloroplasts. With this model we can further investigate the protein synthesis in chloroplasts, for instance the robustness and adaptability of chloroplast translation under perturbations caused by mutations, translational pausing or the influence of codon-anticodon superwobbling on elongation speed and accuracy.
Exploring Sequencing Datasets
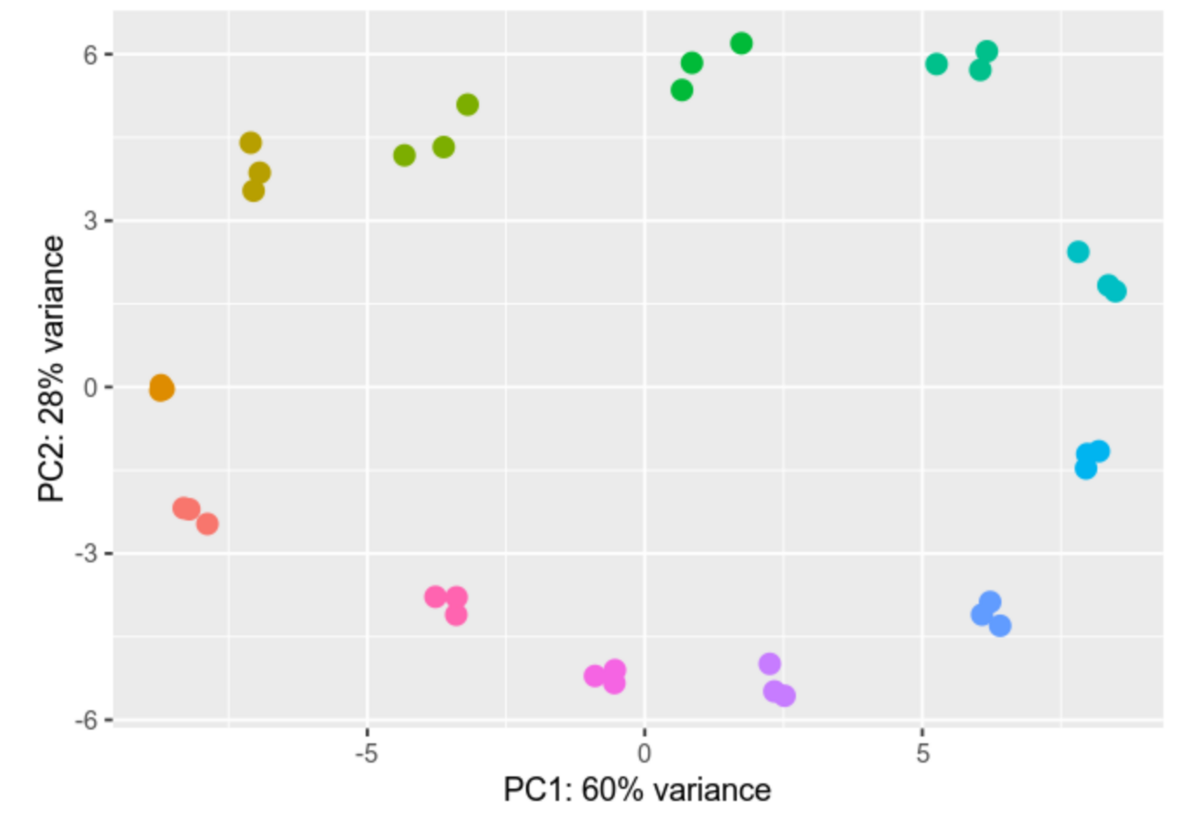
Differential gene expression analysis is a process designed to unravel the intricate web of genes and their varying activity levels across diverse samples or under distinct conditions. We compare gene activity between different cell types, states, or treatments, scrutinizing the nuances of their activation or deactivation and gauging the extent of these changes.
This type of analysis is particularly important for understanding how cells respond to external influences and which biological processes are affected. In practice, differential gene expression analysis can be used for a wide range of applications, such as disease research, the development of new drugs, and the improvement of therapies.
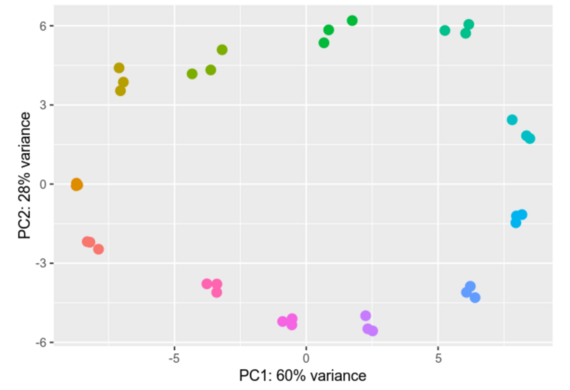
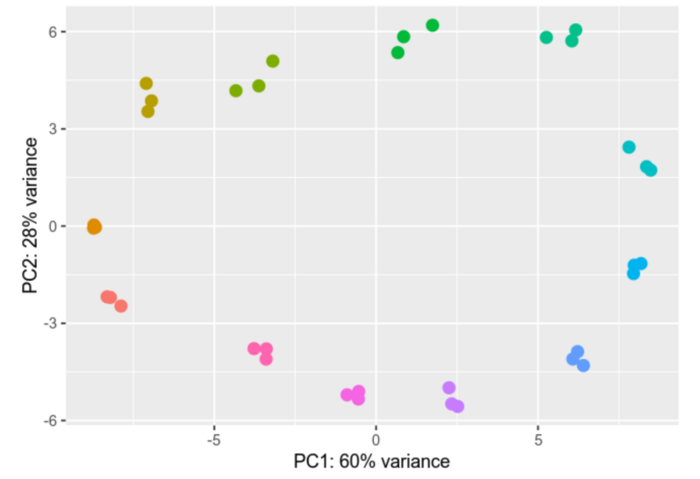
Within our group we analyze Ribosomal profiling (Riboseq) and RNAseq datasets, providing us with valuable insights into differential gene expression. We employ advanced tools like DESeq2 and EdgeR for gene expression analysis. The results of our analyses are depicted through vivid visual representations, including informative heatmaps and Principal Component Analyses (PCAs), effectively illustrating the intricate differences in gene expression profiles.
Protein Complex Formation
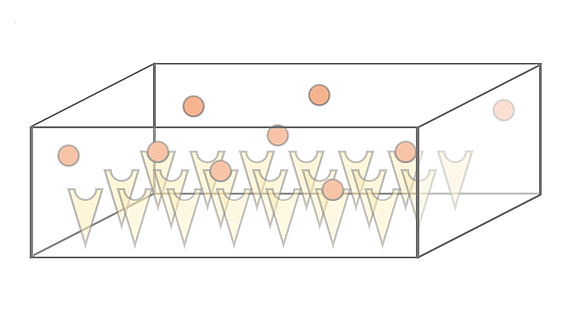

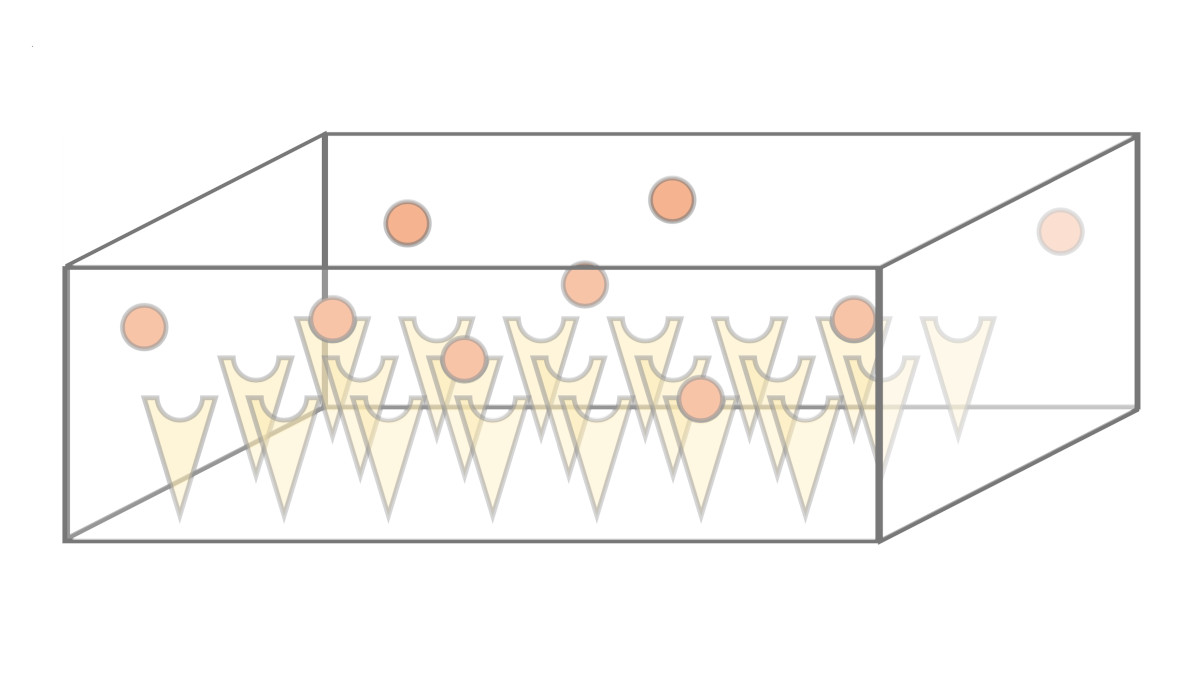
We investigate the dynamics and efficiency of protein complex formation in quasi-twodimensional, silicon-based compartmental expression systems that were introduced by the Bar-Ziv lab (Weizmann Institute, Israel). In these systems, individual protein subunits are synthesized in locally confined domains and are captured on the compartment surface by molecular traps. We systematically study how complex formation and protein-trap binding is governed by the system's parameters, such as synthesis rate, diffusion constant and trap binding affinity of the expressed protein subunits.
Reaction-diffusion Systems with Competing Reactions
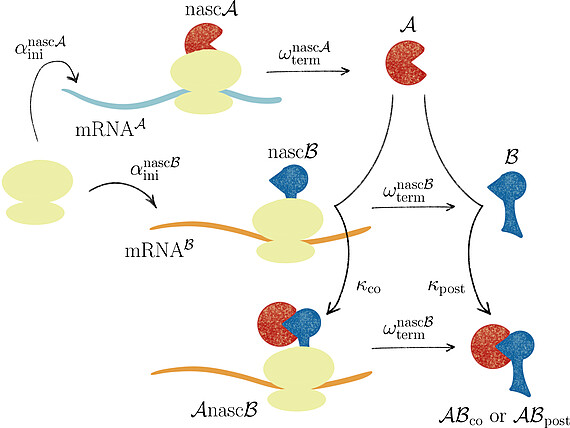
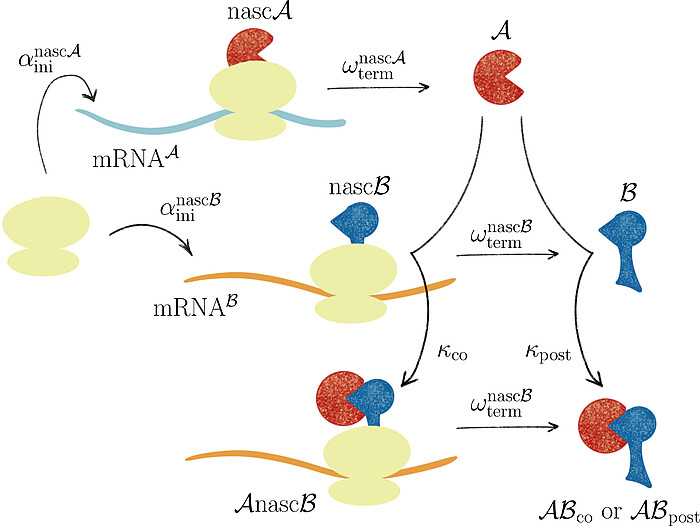
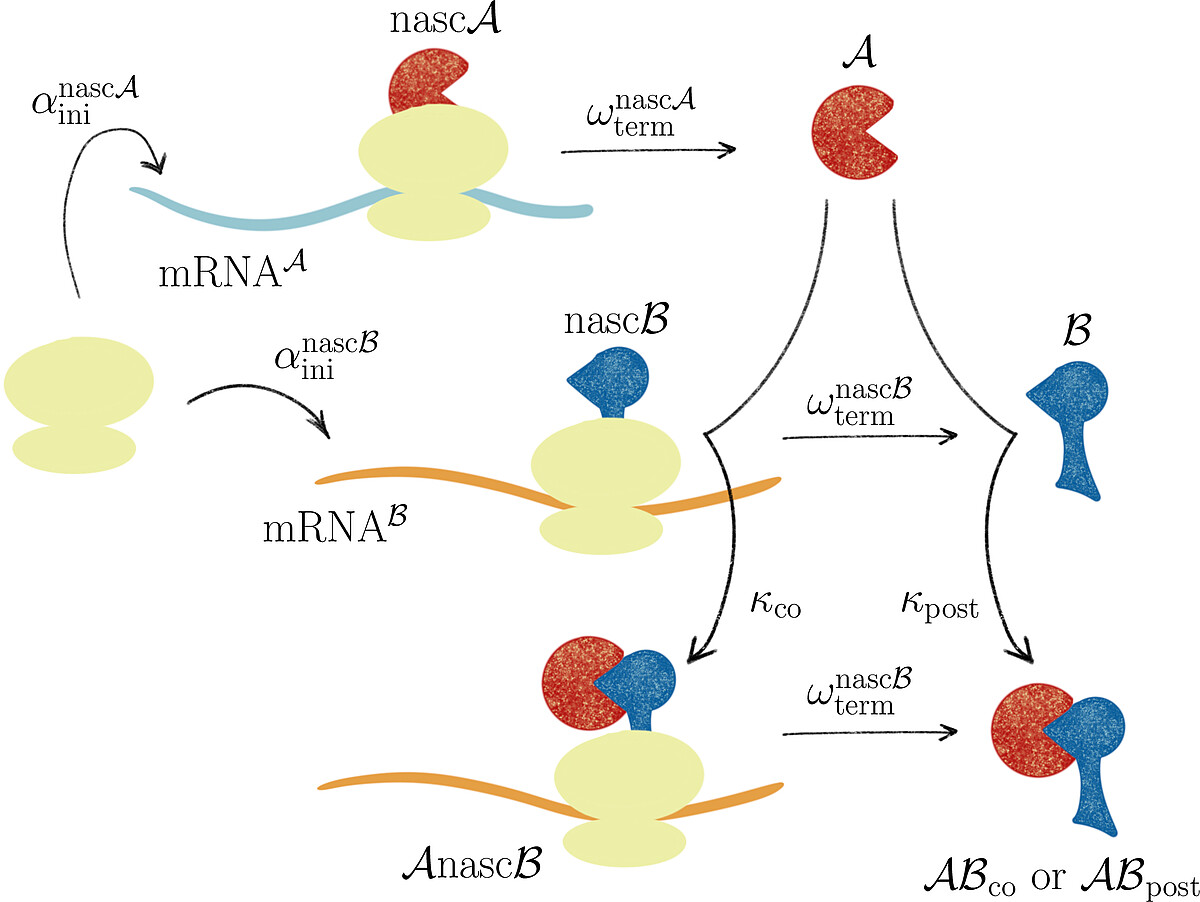
Together with Prof. Johannes Lankeit (Faculty of Mathematics and Physics, LUH), we introduced the concept of asymmetric competing reactions in cellular protein biosynthesis to analyze pathway dominance in co- and post-translational protein complex assembly. Our analysis revealed that the long-term behavior of the system is determined solely by the production rate ratio of the two protein subunits, irrespective of the binding rate constants for post- and co-translational assembly.
Protein Expression Optimisation



The redundancy of the universal genetic code, where 61 different sense codons code for only 20 proteinogenic amino acids, allows for synonymous mutations that do not alter the gene product. Every codon is characterised by different physicochemical properties, which influence for example its tendency to engage in mRNA secondary structure formation and its decoding fidelity. Consequently, different codons are handled at different rates by translating ribosomes. Thus, one can engineer synonymous mRNAs that differ in their local and global translation dynamics.
We developed the software OCTOPOS, which is a tool for computational codon optimisation that proposes synonymous sequences for enhanced heterologous protein expression based on a multitude of mRNA-specific properties. OCTOPOS integrates three levels of modelling:
- a comprehensive, mechanistic mathematical model of in-vivo mRNA translation that considers a broad variety of molecular interaction pathways and their kinetics
- stochastic computer simulations that capture premature ribosome drop-off, mRNA stability, and ribosome jam generation
- a machine learning algorithm that is trained in a host organism-specific manner using large gene expression data sets to take into account and assess the importance of further mRNA-specific features.
Stochastic Modelling of Protein Synthesis
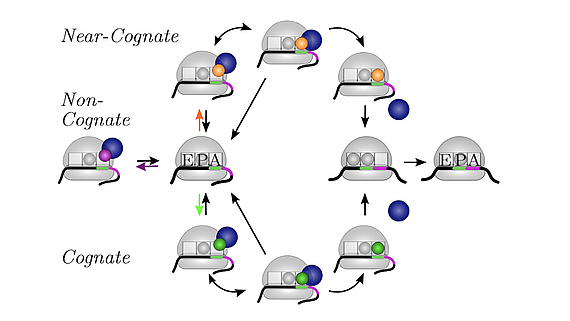
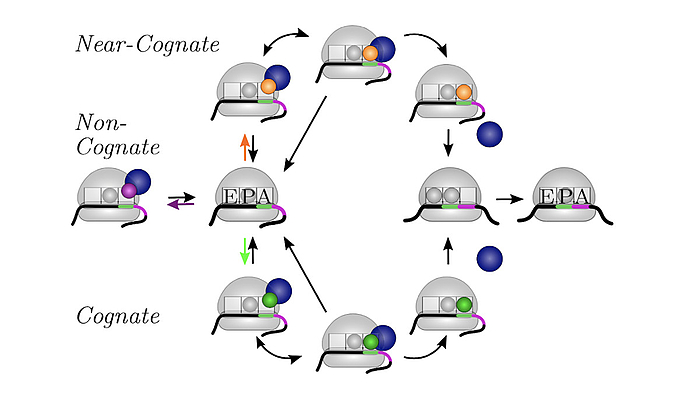
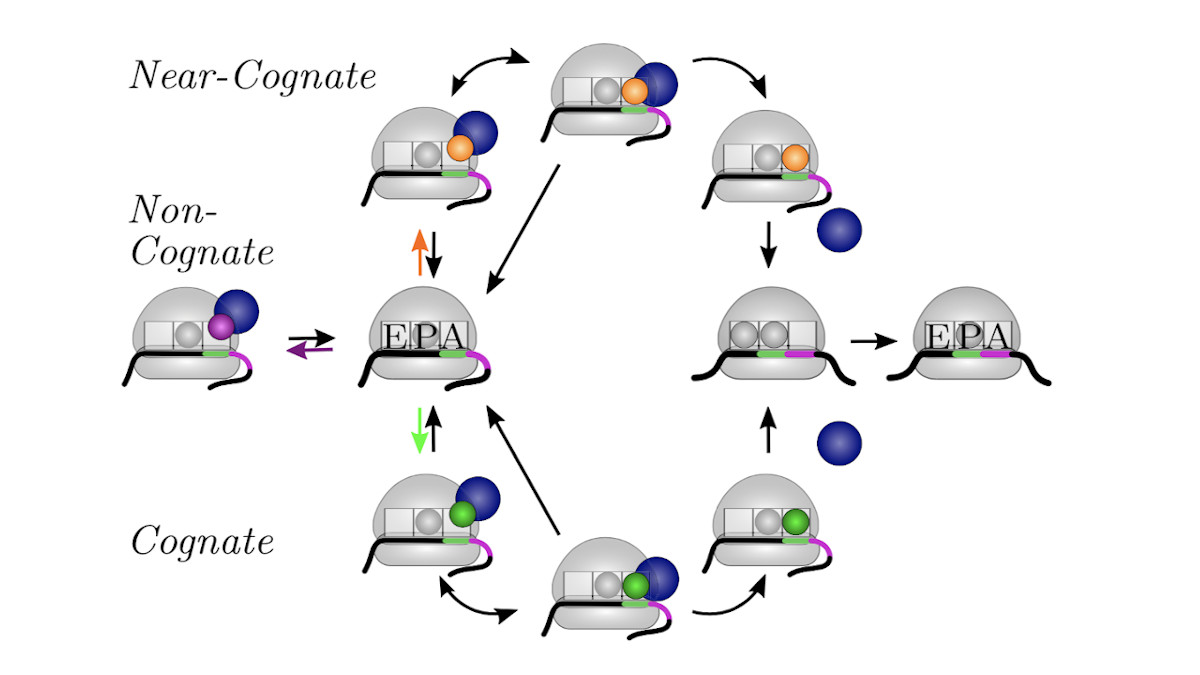
We develop and apply stochastic descriptions of cellular protein synthesis to understand how the speed and fidelity of this process are determined. We use the in-vivo model to determine the effect of disturbances. For example, we examined under which conditions a mild inhibition of elongation factor EF-Tu stalls protein synthesis. Furthermore, we developed a method to infer the kinetics of protein synthesis in the cell from in-vitro measurements.
Stochastic Modelling of Translation in Human Mitiochondria
Mitochondria are organelles in which cellular respiration takes place. They have their own genome and a translation apparatus. We want to describe translation in human mitochondria with the help of our stochastic model and investigate the influence of different mutations in the mitochondrial genome on protein synthesis. The aim is to better understand the mechanisms behind different mitochondriopathies.
Modelling of Photosynthetic State Transitions
State Transitions play an important role in regulating photosynthetic activity under low light conditions in higher land plants. Light harvesting complexes can migrate between the two photosystems to balance absorption energy. Based on the work by Matuszyńska et al. , we describe the mechanism of state transitions by a system of differential equations. Our simulation results are compared to experimental data from the reasearch group plant physiology (Institute for Botany, LUH) to improve the current understanding of the state transition mechanism.
Position-dependent Translation Kinetics
Ribosomes consist of a small and large subunit. During protein synthesis, the nascent peptide chain traverses the ribosomal exit tunnel, an internal tunnel contained in the large subunit (see Fig. 1A). To investigate the interactions between the tunnel and the nascent peptide as well as their effect on the synthesis process, an in-vitro method to monitor the co-translational movement was developed in collaboration with the Rodnina Lab at the Max Planck Institute for Biophysical Chemistry in Göttingen.
A fluorophore was attached to the nascent peptide and the fluorescence signal was detected over time. Co-translational events (e.g. peptide folding) and the structure and chemical environment of the exit tunnel influence the fluorescence signal of the translated mRNA which results in a distinct time-dependent fluorescence signature for that sequence (see Fig. 1B).
The experiment is performed as a bulk experiment and the individual fluorescence signatures superpose to a total fluorescence signature which needs to be decomposed to reveal the underlying information (see Fig. 1C). To gain insights on the position-dependent intensities and the kinetics of the process, we model translation as a Markov process including detailed biological sub-processes of the translation elongation cycle. With this method we discovered, that translation of poly(U) mRNAs dramatically slows down at the fourth UUU codon.
Our method provides a novel approach to study translational kinetics in bulk experiments and can be expanded for the investigations of other biochemical processes driven by processive enzymes. A key concern in our work is to find a meaningful computational analysis of the provided data and to avoid overfitting. Especially for highly complex processes, the number of parameters in the model needs to be carefully defined. On the one hand the complexity of the biochemical process needs to be reflected, on the other hand the results need to be distinct and reliable.
Cooperations
Contact


Prof. Dr. rer. nat. Sophia Rudorf
Phone
Address
115/4160, Herrenhäuser Str. 2
30419 Hannover
30419 Hannover
Building
Room
Prof. Dr. rer. nat. Sophia Rudorf





